This book provides practical information to enable compliance with computer system validation requirements, while highlighting and integrating the Annex 11 guidelines into the computer validation program. The ideas presented in the book are based the author’s experience in the US Department of Defense and regulated industries in various computer systems development, maintenance, and quality functions. A practical approach is presented to increase efficiency and to ensure that software development and maintenance is achieved correctly.
March 27, 2015 Forthcoming by CRC Press
Professional – 368 Pages – 22 B/W Illustrations
ISBN 9781482243628 – CAT# K23442
Montreal, Canada / Ravensburg, Germany, April 11th, 2014
Novatek is delighted to announce a strategic partnership with comes compliance services (CCS). comes compliance services is a European based, highly experienced consulting company specializing in simplifying and streamlining EMA, FDA, ICH, GHTF quality systems, and fulfilling validation and regulatory compliance requirements.
This partnership will allow comes compliance services to continue to expand its excellent compliance services by utilizing Novatek as partner to provide Quality & Compliance services throughout Europe especially to the German-speaking region. The two firms compliment one another’s solutions and service offerings and will work together to provide clients with excellent services in cGMP Compliance, Project Management, Quality Systems Improvement and Validation.
About Novatek International
Established in 1996, Novatek International (www.ntint.com) is leading provider of regulatory compliant software solutions for the Pharmaceutical Industry.
Novatek’s products are business ready, off the shelf and modular providing individual expert solutions with the flexibility of full integration to Nova-LIMS productivity package.
The flagship products Nova-EM for Environmental Monitoring and Nova-Stability for pharmaceutical stability testing have been industry standard for over 10years. Novatek Software has consistently met the requirements of Pharmaceutical Companies and Regulatory Agencies globally. Novatek products deliver value through reducing product time to market, improving process efficiencies, reducing the risks of product contamination and exposure from Regulatory Agencies.
Ermatingen, Switzerland / Ravensburg, Germany, March 17th, 2014
Mediantis Consult GmbH and CCS are delighted to announce the strategic alliance between both consultancy service providers to the regulated industries.
Both companies are providing professional services covering Best Practice approaches of modern quality management and value-driven consulting.
The core capabilities of both companies complement perfectly one another, focusing on GCP & Medical Device quality concepts and GMP and application service platforms.
Mediantis Consult GmbH, managed by Dr. Sabine Schäfer-Preuss and Dr. Iris Teutsch and the CCS network resources, managed by Mr. Markus Roemer, are thereby enabled by a comprehensive skills and expertise set to provide excellent services to their clients.
This extraordinary partnership will allow both providers to continue to expand their excellent services ranges and areas by utilizing each other as partner to provide compliance services throughout Europe.
The two firms complement one another’s service offerings and will work together to provide clients with excellent services in GMP & GCP compliance, Quality Management Systems, Inspection Readiness Programs, Audit Management, Training, and Validation.
About Mediantis Consult GmbH
Mediantis Consult GmbH provides high value professional consulting services for Pharma, Biotech and Medical Device Industries:
Comprehensive CONSULTING in Quality Management Systems tailored to client’s individual needs
Detailed ASSESSMENT of existing Processes with a strong focus on regulatory compliance
Customized and sustainable TRAINING for Quality Manangement Systems and process end users
Development and delivery of innovative EDUCATION programs
Support in CLINICAL DEVELOPMENT with study management, study specific training and medical writing activities
comes compliance services is a premier consulting company specializing in simplifying and streamlining quality systems, and fulfilling validation and regulatory compliance requirements. The services and products are available to GMP, GDP, GLP, GCP and medical device regulated companies and suppliers. CCS is based on a global and comprehensive network setup and can deliver dedicated subject matter experts to different fields and subjects:
Quality Management Services / Pharmaceutical Quality Systems
Comprehensive and risk-based compliance concepts and services
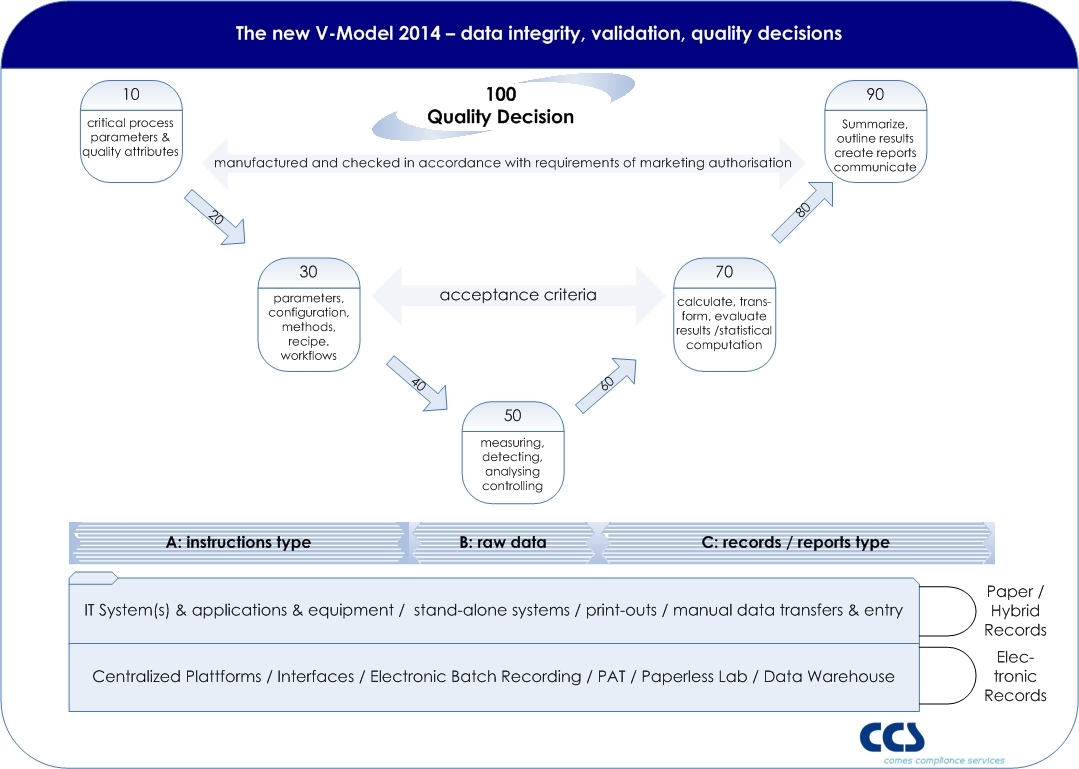
The classical V-model is very often used as the a conceptual model for validation. Most of the V-models are purely designed on a system-based approach, showing in the left wing all sorts of specifications, from URS to design specifications and the right wing all related test phases. Moving away from such a system-based approach in order to follow a risk based approach towards the quality decision making process might impact the existing V-model and a new approach should be used (click picture to enlarge).
click to enlarge
As EU GMP Annex 11 states that applications, and not computerized systems, should be validated, and in real terms we do not validate a computer or system itself, the objective of validation is focused to the quality decision, which is based on the information (recorded in reports and records) derived or given by the entire process, whereas several systems or equipment might be relevant process elements.
EU GMP Chapter 4 – documentation – defines the so called instructions and records/report type. Also the term raw data is used. A GMP record contains such raw data, meta data, master data, on which a decision maker (e.g. Qualified Person, Quality Assurance) is making a quality decision (e.g. Batch Release).
Based on the IT basis such records might support electronic records or is still based on paper records. Ideally all steps (from 10 to 100) are interconnected by the qualified IT architecture.
The fundamental risk is that a wrong quality decision would be made on the basis of insufficient, faulty, tampered or missing data, directly impacting product quality, data integrity, and patient safety. So each system providing data to a GMP relevant record (e.g. Certificate of Analysis, Batch Records, etc.) should be seen as a data sources, forcing, collecting, monitoring, analyzing or calculating data along the entire data flow.
Potentially the highest risk is in each manual step, where data is entered by an operator – whereas a qualified interface of e.g. scanners are reducing the risk of faulty data entries (steps 20, 40, 60, 80).
Step 10 defines critical process parameters, which are derived from product critical attributes, linking product (license) to the process. If a Quality by Design approach is followed, the Design Space will be the basis for the Control Strategy.
A modern approach to computer system validation requires the mapping of the data objects and flows to the final documentation in GMP records. This approach should include the verification of data integrity, as required by EU GMP Annex 11 and US-FDA 21 CFR Part 11.
On the other hand there must be a comprehensive IT strategy including systems, platforms, and interface technology in order to reach the goal of a full electronic recording of all relevant processes. By the way, companies implementing such a holistic data management approach increase efficiency and productivity extensively: Implementing GMP datability & compliance as guarantor of success.
For more information contact us at: talk@comes-services.com or use our online contact form.
Aus unserer Sicht ist die aktuelle Draft Version, die zur Kommentierung veröffentlich wurde, sehr undurchsichtig und unzureichend formuliert. Auch die Referenzen zur QWP Guideline und ICH Q11 und weitere zeigen erhebliche Inkonsitenzen und unklare Definitionen und Festlegungen.
So unterscheiden sich alleinig schon die Festlegungen, was zu einem Prozess und Lebenszyklus gehört, die unterschiedlichen Definitionen, sogar im Anhang 15 selbst. Zur Qualifzierung und Validierung wurde dann noch der Schritt der Verifizierung aufgenommen, ohne dies näher zu beschreiben. Es ist sicherlich empfehlenswert, dass zu allererst eine Festlegung der Definition eines Prozesses selbst erfolgen sollte. Die QWP Guideline beinhaltet beispielsweise im Anhang 1 ein sogenanntes Process Validation Scheme, das sehr sinnvoll ist, aber im Anhang 15 nicht direkt genannt wird. Der ICH Q11 gbit eine Control Strategy vor, die in einem Dokument “manufacturing process development” definiert sein soll.
In Kapitel 3.2 wird beispielsweise definiert, dass eine URS (für new facilities, systems or equipment) erzeugt werden muss, und dass die URS “should be a point of reference throughout the validation life cycle”. Dass aber die Anforderungen aus einer Prozessspezifikation erzeugt werden müssen, und demzufolge alle Prozesselemente (ob als System, Equipment, Gebäude, etc. definiert wären) entsprechend qualifiziert sein müssen, ist nur unzureichend formuliert. Als Lebenszyklus findet man diesen auf Produkte, Prozesse, Validierung und natürlich Daten. Ob dieses Dokument dann VMP genannt wird, und ob dies “pro Site” erzeugt wird, sollte nicht als eine Anforderung in den Annex 15 münden. Interessant könnten aber entsprechende Prozesse sein, die von externen Firmen (outsourced activities – Kapitel 7 EU GMP) durchgeführt werden. Die aktuelle Draft Version des Anhang 16 fordert hierfür ein “comprehensive diagram”, das ebenso als Grundlage zur Prozessvalidierung gelten sollte.
Ob nun einzelne Prozessschritte wie Reinigung und Transport, die natürlich wichtig sind, einzeln genannt werden müssen, auch noch in der Verbindung des Worts “Verification”, sollte diskutiert werden. Prozesse bestehen aus Prozesselementen und Prozesschritten, eigentlich über den gesamten Warenfluss (Ausgangsstoffe bis zum Fertigarzeimittel). Daher sollten Prozessbeschreibungen den gesamten Material-, Personal- und Datenfluss beinhalten, in den entsprechenden Umgebungen und Vorbedingungen.
Die aktuelle Draft Version beihaltet viele Punkte zur Diskussion und Bewertung. Daher nochmals unsere Empfehlung, damit die EMA Arbeitsgruppe die aktuelle Version verbessern kann:
Nutzen Sie unbedingt die Chance die Draft Version des Anhang 15 zu kommentierten.
Erfolgreicher erster QPmeetsIT Dialog initiiert durch comes compliance services und Systec & Services.
Am 4. Februar 2014 trafen sich zahlreiche Experten zum ersten QPmeetsIT Dialog im Karlsruher Schlosshotel. Als Medienpartner konnte der Editio Cantor Verlag (ecv) für die Veranstaltung gewonnen werden.
Moderne Qualitätsphilosophien propagieren einen Paradigmenwechsel hin zu kontinuierlichen Verbesserungen durch schlanke Qualitätssysteme und fundiertem Qualitätsrisikomanagement. Aber wie setzt man diese Vision wirklich in die Tat um?
Dieser Frage gingen die vier Referenten Dr. Bernd Renger, Markus Roemer, Lukas Eberle und Titus Krauss zusammen mit den Teilnehmern nach.
Dr. Renger konnte aufzeigen, dass zwingend Handlungsbedarf besteht, da die neu gefassten Regularien (Annex 16 aber auch der bestehende Annex 11) zwingend die Einbeziehung von Software-Lösungen fordert. Hier sieht er die Sachkundige Person (Qualified Person) mit in der Verantwortung. Markus Roemer stellt dar, wie sich diese sich verändernden Regularien auf die Validierungsansätze und die Validierungsmethoden auswirkt. Beides untermauerte er mit praxisnahen Beispielen.
Am Nachmittag zeigte Titus Krauss, wie die Anforderungen mit methodenbasieren Datenmodellierungen sowie Software-Lösungen bereits heute erfüllt werden können. Anhand einer Live-Demo konnten sich die Teilnehmer ein Bild der von der Lösung machen. Im Anschluss konnte Lukas Eberle live vorführen, dass F. Hoffmann-La Roche diese Methoden und Software-Lösungen bereits praktisch im Einsatz hat.
Die Resonanz der Teilnehmer war durchweg positiv. Alle Teilnehmer regten an, diese Dialogreihe fortzusetzen. comes compliance services und Systec & Services werden diesem Wunsch gerne entsprechen.
Mobile applications (apps) can help people manage their own health and wellness, promote healthy living, and gain access to useful information when and where they need it. These tools are being adopted almost as quickly as they can be developed. According to industry estimates, 500 million smartphone users worldwide will be using a health care application by 2015, and by 2018, 50 percent of the more than 3.4 billion smartphone and tablet users will have downloaded mobile health applications.
Mobile apps are software programs that run on smartphones and other mobile communication devices. Mobile medical apps are medical devices that are mobile apps, meet the definition of a medical device and are an accessory to a regulated medical device or transform a mobile platform into a regulated medical device.
Apps can be used in several areas and for different objectives. Consumers can use both mobile medical apps and mobile apps to manage their own health and wellness, such as to monitor their caloric intake for healthy weight maintenance. Other apps aim to help health care professionals improve and facilitate patient care, are used in combination with Blood Establishment Computer Software (BECS), may support GCP case report forms (CRF), or are a verification tool for falsified products (GMP & GDP).
In any case such apps must be validated according best practice (e.g. ISPE GAMP 5) and related quality standards. CCS is one of the few and leading compliance consulting firms supporting the design, development, operation, and validation of mobile apps, which are used as medical devices or in any GXP related area / function.
Erfolgreiche Unternehmen zeichnen sich dadurch aus, dass sie über eine positive Wissensbilanz und eine gelebte Informationskultur verfügen. Auf dieser Grundlage können Entscheidungen getroffen und nachhaltige Lösungen und Konzepte entwickelt werden, die einen wesentlichen Marktvorteil bedeuten können. Dabei entsteht das Wissen aus Erfahrung und Information; die Information wiederum wird aus Daten und Kommunikation erhalten; gefiltert aus variablem Daten- und (statistischer) Prozesskontrolle und durch ein Risikomanagement.
Aber ist das wirklich die Realität?
Moderne Qualitätsphilosophien propagieren einen Paradigmenwechsel hin zu kontinuierlichen Verbesserungen durch schlanke Qualitätssysteme und fundiertem Qualitätsrisikomanagement. Aber wie setzt man diese Vision wirklich in die Tat um?
Wie funktioniert ein prospektives Risikomanagement mit akzeptablen Restrisiko-Festlegungen und wie gelingt eine gemeinsam unternehmensweite Informationsintegration. Nur aus Fehlern zu lernen („das passiert uns kein zweites Mal“) ist ineffizient und nicht akzeptabel; der richtige Zeitpunkt, wann die Information vorhanden ist (“hätten wir das früher gewusst”), ist ebenso entscheidend.
Moderne Qualitätssysteme und Methoden machen Erfolge planbar und transparent und liefern bessere Entscheidungsgrundlagen: Weniger Fehlentscheidungen treffen und ein akzeptables Restrisiko sicherstellen. Hierfür werden interdisziplinäre Problemlösungen und entsprechende Lösungskonzepte benötigt – informieren Sie sich und tauschen Sie sich aus beim QP>>meets<<IT Dialog mit den folgenden Themen und Lösungen:
Wissensmanagement, Quality Risk Management (QRM) und GMP Daten-Mapping zur effizienten und elektronischen Erzeugung von Quality Reports (PQR, APR) und risikobasierte Methoden und Grundlagen für Quality Decisions (Batch Certification, OOS, Abweichungen);
Validierung und Sicherstellung der Datenintegrität, Definition von Rohdaten und GMP relevanten Aufzeichnungen;
Verwendung von elektronischen Informationen und elektronische Signaturen;
Rollen und Verantwortlichkeiten im Pharmazeutischen Qualitätssystem (Update: EU GMP Kapitel 2);
Qualifizierte Infrastruktur und strategisches IT Programm (Compliance, Security, Governance, “Best Practice”);
Mit “Live-Demo” eines Data-Mining Ansatzes und Praxis-Erfahrungsbericht!
Powered by:
>>
Systec & Services GmbH und comes compliance services
We are very often asked about the retrospective validation approach. So this short article below (PDF download) describes some basics and careful considerations about this special topic and related concepts and interpretations, with making no claim to be complete.